



Ces dernières années, une grande attention a été accordée à l’utilisation des antimicrobiens en médecine vétérinaire. L’utilisation responsable des antibiotiques est essentielle au fonctionnement optimal du médicament, en limitant la résistance et en assurant la sécurité alimentaire. En 2014, Dopharma a publié plusieurs fois dans son bulletin d’information un article contenant des informations pratiques sur les antibiotiques. Ces articles sont fusionnés en un seul document, qui est également disponible pour vous entéléchargement (pdf, anglais), mais vous pouvez également lire le texte ci-dessous.
Adopter une approche raisonnée de l’antibiothérapie, ce n’est pas simplement réduire le nombre de prescriptions. Cela implique surtout de choisir la molécule la plus adaptée à la situation, en s’appuyant sur des outils comme l’antibiogramme ou les référentiels spécifiques à chaque espèce animale. Selon la sensibilité bactérienne, différents principes actifs restent envisageables, même en respectant l’ordre de priorité (1er, 2e, 3e choix). Connaître la pharmacodynamie et la pharmacocinétique devient alors indispensable pour faire un choix solide. Même en dehors des indications officielles, dans le cadre des bonnes pratiques vétérinaires, ces éléments permettent de justifier scientifiquement la démarche.
A voir aussi : Liste fromage pour diabétique : quelles portions au quotidien ?
Intro
Pharmacodynamique et pharmacocinétique
La pharmacodynamique décrit l’action d’un médicament sur l’organisme. Les antibiotiques, pour leur part, sont classés selon leur mode d’action et leurs principales propriétés. Le spectre d’activité constitue aussi un critère clé, même s’il n’est pas traité en détail ici. De son côté, la pharmacocinétique s’intéresse à la façon dont le corps gère le médicament, avec quatre étapes : Absorption, Distribution, Métabolisme, Élimination (AMDE).
Parmi les paramètres majeurs à surveiller : Cmax (concentration maximale), Tmax (délai pour atteindre cette concentration), CL (clairance), T1/2EL (demi-vie d’élimination), et Vd (volume de distribution). Ces valeurs varient en fonction du médicament, de sa formulation, du mode d’administration et de la dose prescrite.
A voir aussi : Apaiser la douleur des ganglions inguinales en attendant le médecin
Bactéricide ou bactériostatique
On connaît la distinction : les antibiotiques bactéricides éliminent les bactéries, tandis que les bactériostatiques freinent leur croissance. Mais en laboratoire, la frontière n’est pas si nette : certains bactériostatiques, à doses élevées, tuent aussi les bactéries, et tous les bactéricides ne sont pas systématiquement létaux dans le délai imparti. En pratique, on considère souvent que les bactéricides sont préférables lors d’infections aiguës ou quand l’immunité est défaillante. Cependant, il existe des situations où les bactériostatiques sont plus indiqués : par exemple, lorsque la destruction rapide de bactéries libérant des endotoxines peut aggraver le tableau clinique.
Constante de lipophilité et de dissociation
Pour atteindre leur cible, les antibiotiques doivent pénétrer dans les tissus et franchir les membranes cellulaires. Outre le transport actif et la diffusion à travers les pores, la diffusion passive dépend fortement de la lipophilie et du pKa de la molécule. Lorsqu’une substance est très lipophile, elle traverse plus aisément la bicouche lipidique des membranes, atteignant ainsi des concentrations efficaces dans des compartiments comme la synovie, l’œil ou le liquide céphalo-rachidien. Pour les substances modérément lipophiles, la distribution dépend du degré de liaison aux protéines plasmatiques et donc du volume de distribution.
Le pKa d’un médicament indique la proportion de la forme ionisée et non ionisée. Seule la forme non ionisée franchit les membranes par diffusion passive. Si le pH varie d’un tissu à l’autre (par exemple entre le sang et le tissu pulmonaire), l’antibiotique s’accumule là où il est le plus ionisé. Les bases faibles s’accumulent dans les milieux plus acides, les acides faibles dans les milieux plus basiques : c’est le phénomène de piégeage ionique. Ce principe est particulièrement notable entre le plasma et certains tissus (poumon, prostate, lait, urine des carnivores ou des herbivores). Ainsi, pour obtenir des concentrations élevées dans les poumons, mieux vaut choisir un antibiotique lipophile avec un pKa élevé.
Concentration ou effet dépendant du temps
Certains antibiotiques agissent mieux quand leur concentration dépasse largement la CMI (concentration minimale inhibitrice) : plus la dose crête est élevée, plus l’élimination bactérienne est rapide et efficace.
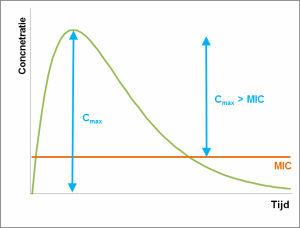
Dans ce cas, la différence entre la concentration maximale dans le plasma (Cmax) et la CMI est décisive. Pour un résultat optimal, la Cmax devrait être dix fois supérieure à la CMI. On peut, par exemple, obtenir ce pic par des doses ponctuelles, plutôt qu’une administration continue. Ici, la durée du maintien de la concentration élevée compte moins, car ces molécules disposent généralement d’un effet post-antibiotique marqué.
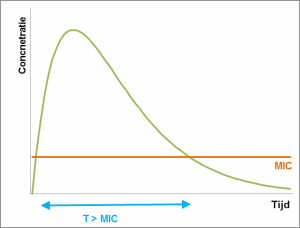
Pour d’autres familles, l’efficacité dépend du temps pendant lequel la concentration reste au-dessus de la CMI (T>CMI). Ici, l’objectif est de maintenir la concentration au-dessus de la CMI pendant au moins la moitié de l’intervalle entre deux doses. Si la concentration reste suffisante pendant 6 heures, il faudra espacer les prises de 12 heures. Dans ces cas, il est préférable de répartir les prises sur la journée pour une exposition continue.
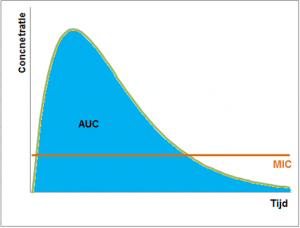
Il existe aussi des antibiotiques qui cumulent ces deux exigences : atteindre un pic élevé et le maintenir suffisamment longtemps. Ici, on se réfère à l’ASC (aire sous la courbe) : l’ASC/CMI doit dépasser 125 pour garantir une efficacité optimale.
Pénicillines
Pharmacodynamique
Les pénicillines exercent une action bactéricide en bloquant la synthèse du peptidoglycane de la paroi cellulaire bactérienne. Elles se fixent de manière irréversible aux protéines de liaison à la pénicilline (PBP), empêchant l’assemblage du peptidoglycane, indispensable à la division cellulaire. Cet effet se produit uniquement lors de la phase de division, lorsque la paroi est en construction.
On distingue clairement les pénicillines à spectre étroit, efficaces surtout contre les bactéries Gram positif, et celles à large spectre qui agissent aussi sur les Gram négatif. Cela provient en partie de la structure bactérienne : chez les Gram positif, le peptidoglycane est exposé, ce qui rend les PBP accessibles. Chez les Gram négatif, une couche lipidique supplémentaire protège la paroi, compliquant l’accès aux PBP. Les pénicillines à spectre étroit franchissent mal cette barrière, contrairement aux molécules à large spectre. D’autres éléments, comme la forme des PBP, la quantité de peptidoglycane ou la production d’enzymes β-lactamases, modulent aussi la sensibilité à ces antibiotiques.
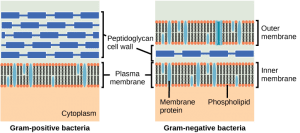
Les pénicillines restent sans effet sur les bactéries dépourvues de paroi cellulaire, comme les Mycoplasma spp. Cette spécificité explique aussi leur excellente tolérance chez l’animal, qui ne possède pas de PBP.
Constante de lipophilité et de dissociation
Les pénicillines présentent une faible lipophilie et un volume de distribution restreint. Leur biodisponibilité orale reste donc limitée, à l’exception de la pénicilline V, résistante à l’acidité gastrique. Leur faible pKa limite leur diffusion membranaire : elles circulent majoritairement sous forme ionisée dans le plasma, franchissant difficilement les membranes. Par exemple, leur concentration dans la mamelle ne représente qu’un cinquième de celle du plasma. Lors d’une inflammation, cependant, la baisse du pH tissulaire favorise la forme non ionisée, facilitant la pénétration et permettant d’atteindre des concentrations efficaces dans des tissus normalement peu accessibles.
Concentration ou effet dépendant du temps
Les pénicillines agissent selon un mécanisme dépendant du temps : leur efficacité augmente avec la durée d’exposition de la bactérie à l’antibiotique. Des administrations fractionnées ou l’usage de formes à libération prolongée (comme la procaïne) permettent de maintenir une concentration suffisante sur la durée.
Chez certaines bactéries, notamment les entérocoques, une concentration trop élevée peut paradoxalement diminuer l’efficacité (effet Eagle), soulignant l’importance d’adapter le schéma posologique à la nature du pathogène.
Combinaison d’antibiotiques
Les pénicillines n’agissent que sur les bactéries en division. Il est donc déconseillé de les associer à des bactériostatiques qui bloquent la croissance. Exception notable : la combinaison avec les aminoglycosides, qui sont eux-mêmes bactéricides. L’association β-lactamines/aminoglycosides potentialise l’action des deux, car la paroi fragilisée par la pénicilline facilite la pénétration de l’aminoglycoside.
Résistance
La résistance aux pénicillines s’explique principalement par la production de β-lactamases, des enzymes qui dégradent le noyau β-lactame et empêchent la fixation aux PBP. Ce mécanisme, souvent porté par des plasmides, est particulièrement courant chez les bactéries Gram négatif, favorisant la diversité des β-lactamases chez ces dernières. Ainsi, les pénicillines à spectre étroit rencontrent peu de résistance chez les Gram positif, tandis que les molécules à large spectre voient la résistance émerger plus rapidement chez les Gram négatif.
Les groupes de bactéries résistantes aux pénicillines incluent le SARM (Staphylococcus aureus résistant à la méthicilline), les bactéries productrices d’ESBL (β-lactamases à spectre élargi) et celles produisant de l’AMPC. Ces mécanismes rendent ces bactéries insensibles, non seulement aux pénicillines, mais souvent à plusieurs autres familles de β-lactamines. Certaines pénicillines, comme la cloxacilline, sont conçues pour résister à l’action des β-lactamases. D’autres solutions consistent à associer les β-lactamines à des inhibiteurs enzymatiques, comme l’acide clavulanique.
Enfin, d’autres mécanismes de résistance existent : modification des PBP, réduction de la perméabilité à l’antibiotique ou augmentation de l’efflux. La diminution du nombre de pores membranaires, critique pour le passage de la molécule, touche surtout les Gram négatif comme E. coli.
Céphalosporines, polymyxines et fluoroquinolones
Pharmacodynamique
Les céphalosporines et les polymyxines ciblent la paroi cellulaire des bactéries et agissent toutes deux comme bactéricides. Les céphalosporines, proches des pénicillines, bloquent la synthèse du peptidoglycane en se fixant aux PBP. Les molécules de première et deuxième génération (céphalexine, céphalonium, cefapirine) sont surtout actives sur les Gram positif, tandis que les générations plus récentes (ceftiofur, cefquinome) élargissent leur spectre aux Gram négatif.
Les polymyxines, quant à elles, perturbent la membrane externe des Gram négatif en se liant aux lipopolysaccharides (LPS), ce qui désorganise la membrane et conduit à la mort bactérienne. Ces LPS étant aussi des endotoxines, leur neutralisation par les polymyxines constitue un atout supplémentaire.
L’efficacité des polymyxines peut toutefois être réduite en présence d’ions calcium ou magnésium, qui se lient également aux LPS. Les fluoroquinolones, elles, bloquent la synthèse de l’ADN bactérien en inhibant l’ADN gyrase et la topoisomérase, deux enzymes indispensables à la réplication et à la réparation de l’ADN. Ce mécanisme entraîne, en plus, la formation de cassures d’ADN et un stress oxydatif létal pour la bactérie.
Le spectre d’activité des fluoroquinolones varie selon la molécule. Par exemple, la fluméquine cible principalement les entérobactéries (E. coli, Salmonella), tandis que l’enrofloxacine, grâce à l’ajout d’un atome de fluor, agit aussi sur certains Gram positif et anaérobies, élargissant ainsi les indications cliniques.
Constante de lipophilité et de dissociation
Les céphalosporines et polymyxines sont peu lipophiles et se distribuent dans un volume limité, d’où une mauvaise absorption digestive. Elles sont donc surtout administrées par voie parentérale (injection, intramammaire) ou, pour la colistine, par voie orale pour cibler le tractus digestif. Leur faible pKa favorise leur accumulation dans les tissus où le pH est plus élevé que le plasma pour les céphalosporines, ou plus bas pour les polymyxines.
Les fluoroquinolones de dernière génération, à l’image de l’enrofloxacine, se distinguent par leur lipophilie élevée et leur faible fixation aux protéines plasmatiques. Résultat : elles diffusent efficacement dans de nombreux tissus, y compris le cerveau ou les voies urinaires, et s’accumulent dans les cellules phagocytaires, ce qui les rend actives sur les bactéries intracellulaires. L’absorption digestive reste très bonne, mais peut être ralentie ou réduite si l’alimentation est riche en ions magnésium ou aluminium.
Les fluoroquinolones sont amphotères, caractérisées par deux pKa, ce qui leur permet de bien diffuser dans des tissus à pH variable.
Concentration ou effet dépendant du temps
Les céphalosporines agissent selon un mode dépendant du temps : il faut maintenir la concentration au-dessus de la CMI aussi longtemps que possible pour une efficacité optimale. L’intervalle entre deux administrations ne doit pas dépasser deux fois la durée pendant laquelle la concentration reste efficace, à ajuster selon l’espèce.
Polymyxines et fluoroquinolones sont des molécules à effet dépendant de la concentration : ce qui compte, c’est d’atteindre une concentration élevée, idéalement dix fois la CMI, au site de l’infection.
Combinaison d’antibiotiques
Comme pour les pénicillines, les céphalosporines ne doivent pas être associées à des bactériostatiques, sauf exception pour les aminoglycosides, avec lesquels un effet synergique a été démontré. Les polymyxines peuvent être associées aux sulfamides et au triméthoprime pour renforcer l’activité sur certaines entérobactéries comme P. aeruginosa. En pratique, l’association amoxicilline/polymyxine est courante pour traiter des infections mixtes et freiner la résistance. Chez l’homme, ce type de combinaison est aussi utilisé contre des bactéries multirésistantes.
Les fluoroquinolones présentent également une synergie lorsqu’elles sont associées aux β-lactamines ou aux aminoglycosides.
Résistance
La résistance aux céphalosporines résulte principalement de la modification des PBP, d’une perméabilité réduite, d’un efflux accru et de l’inactivation enzymatique par des β-lactamases. Les générations récentes résistent mieux à ces enzymes grâce à des modifications structurelles. Aux Pays-Bas, la résistance aux céphalosporines touche surtout les agents de mammite Gram positif chez les bovins ou B. bronchiseptica chez le porc.
La résistance aux polymyxines reste rare, mais elle peut survenir suite à une modification du LPS de la membrane bactérienne, réduisant l’attraction pour la molécule. Des cas de résistance plasmidique ont été détectés chez E. coli isolé d’animaux. Une résistance croisée totale existe entre les différentes polymyxines. La colistine, par exemple, rencontre une résistance chez Salmonella chez les bovins.
Les fluoroquinolones, en inhibant la réparation de l’ADN, favorisent l’apparition de mutations et de résistances. Les mécanismes mis en jeu incluent la modification du site cible, la diminution de la perméabilité membranaire, l’efflux actif et le blindage du site de liaison. La réduction du nombre de pores OMP dans les Gram négatif limite aussi l’entrée de la molécule et, par ricochet, d’autres antibiotiques utilisant ces passages.
Une résistance à une fluoroquinolone entraîne souvent une moindre sensibilité à l’ensemble de la famille, surtout avec les anciennes molécules ou en cas de résistances multiples. Aux Pays-Bas, des résistances à l’enrofloxacine et à la fluméquine sont régulièrement isolées, notamment chez E. coli et Salmonella, tant chez les bovins que chez les volailles et les porcs.
Diaminopyrimidines et sulfonamides
Pharmacodynamique
Les diaminopyrimidines et les sulfamides perturbent la synthèse de l’ADN bactérien en bloquant la production d’acide folique. Les sulfamides entravent la première étape, en concurrençant l’acide para-aminobenzoïque (PABA) pour l’accès à l’enzyme dihydroptéroate synthétase. Les diaminopyrimidines, elles, interviennent plus loin sur la voie, inhibant la dihydrofolate réductase et empêchant la formation du tétrahydrofolate, forme active de l’acide folique.
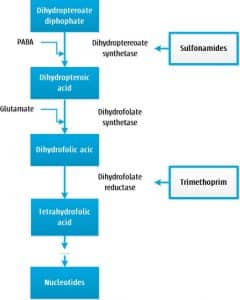
Les sulfamides sont bactériostatiques, les diaminopyrimidines bactéricides à dose suffisante. Leur association devient bactéricide. Toutefois, l’efficacité des sulfamides peut être compromise dans un environnement riche en PABA, comme lors d’une inflammation avec pus ou nécrose, ce qui explique certains échecs cliniques malgré des résultats in vitro encourageants.
Leur spectre est large, car l’acide folique est vital pour la synthèse des acides nucléiques de la plupart des bactéries. Seules quelques entérobactéries peuvent prélever directement l’acide folique de leur environnement, ce qui les rend moins sensibles aux sulfamides.
Chez les mammifères, la synthèse de l’acide folique est également cruciale, notamment pendant la gestation. Même si la dihydrofolate réductase bactérienne est bien plus sensible au triméthoprime que son homologue mammifère, la prudence s’impose chez les femelles gestantes.
Constante de lipophilité et de dissociation
Les sulfamides et diaminopyrimidines affichent une lipophilie modérée. La fixation aux protéines plasmatiques des sulfamides est généralement élevée, mais varie selon la molécule et l’espèce. Les diaminopyrimidines se lient modérément. Les sulfamides diffusent bien dans les tissus, y compris les liquides synovial et céphalo-rachidien, tandis que les diaminopyrimidines pénètrent aussi en intracellulaire grâce à un volume de distribution plus important.
Leur pKa diffère : les sulfamides (pKa faible) s’accumulent dans les tissus à pH acide, les diaminopyrimidines (pKa élevé) dans les tissus à pH plus alcalin.
L’élimination se fait en partie après biotransformation hépatique, puis excrétion par les reins, la bile, le lait ou les fèces. Une fraction est aussi éliminée inchangée dans l’urine, avec des variations dues au pH urinaire. Chez les animaux, la demi-vie varie selon l’espèce et la molécule, influencée par le taux de biotransformation.
L’injection de sulfamides peut être irritante en raison de leur alcalinité. Une administration lente est donc recommandée.
Ratio et demi-vies
Pour les combinaisons triméthoprime/sulfonamide, on vise un ratio in vivo de 1:20, le triméthoprime étant vingt fois plus puissant. Ce rapport s’obtient le plus souvent en administrant un produit dosé à 1:5. Les études montrent que la synergie s’exerce sur une large plage de ratios, de 1:1 à 1:1000, et que les concentrations tissulaires du triméthoprime dépassent celles du plasma grâce à son grand volume de distribution.
Tableau 1 Demi-vie d’élimination du triméthoprime et de divers sulfamides chez les bovins, veaux, porcs et chevaux après injection intraveineuse (heures)
| Bœuf | Veau | Cochon | Cheval | |
| Triméthoprime | 1,0, 2,0 | 1,9, 2,1 | 2,7, 2,9 2* | 2,0, 3,0* |
| Sulfadiazine | 2,5 | 4,4 1 | 2,8 | 4,6 |
| Sulfaméthoxazole | 2,3 | 12,8 * | 12,9 et 12,4 2 | 3,5 |
| Sulfachlorpyridazine | 1,2 | 13,1* | 3,0 4 | 3,8 |
| Sulfadoxine | 10,8, 13,0 | 12,9 | 8,2 et 8,4 3 | 14,2 |
1 : Chez le veau, la demi-vie passe de 5,7 à 3,6 heures entre 1 et 42 jours. 2 : Chez le porc, demi-vie chez les animaux sains et atteints de pneumonie. 3 : Après administration de sulfadoxine seule ou associée au triméthoprime. 4 : Après prise orale chez le porc. * Méthode d’administration non précisée dans l’étude.
La demi-vie des sulfamides varie donc selon la molécule et l’espèce, et n’est pas toujours calée sur celle du triméthoprime. Le ratio optimal n’est donc respecté qu’une partie du temps, mais l’efficacité reste bonne grâce à la synergie large et à la distribution tissulaire.
Concentration ou effet dépendant du temps
Les deux familles agissent selon un mode dépendant du temps : il faut maintenir la concentration au-dessus de la CMI aussi longtemps que possible.
Combinaison d’antibiotiques
Diaminopyrimidines et sulfamides sont fréquemment associés pour obtenir un effet bactéricide. On peut aussi combiner la TMP/S avec d’autres familles afin d’élargir le spectre, par exemple en association avec une pénicilline pour couvrir les bactéries anaérobies et aérobies, même en présence de pus ou de débris.
En revanche, il n’est pas recommandé de dissoudre TMP/S avec d’autres médicaments dans l’eau de boisson : la solubilité du triméthoprime et des sulfamides varie selon le pH, et les excipients ou autres médicaments peuvent perturber cette solubilité.
Les sulfamides ne doivent pas être associés à la procaïne, qui entre en compétition avec eux sur la cible bactérienne.
Résistance
La résistance aux sulfamides se rencontre régulièrement chez E. coli et certaines souches de Salmonella, chez les bovins, porcs et volailles. Plusieurs mécanismes sont impliqués :
Voici les principaux procédés par lesquels les bactéries développent une résistance aux sulfamides :
- Modification de l’enzyme dihydroptéroate synthétase, empêchant la fixation des sulfamides
- Diminution de la perméabilité membranaire, ce qui limite la pénétration de l’antibiotique
- Production accrue de PABA, qui annule la compétition avec le médicament
La résistance croisée entre sulfamides est totale : une souche résistante à l’un sera résistante aux autres. Des cas de multirésistance, incluant le triméthoprime ou la streptomycine, ont été observés, notamment en cas de résistance plasmidique.
La résistance aux diaminopyrimidines repose le plus souvent sur une mutation de la dihydrofolate réductase, rendant l’enzyme insensible, mais aussi sur une diminution de la perméabilité. Ces mécanismes peuvent être d’origine chromosomique ou plasmidique.
Tétracyclines et aminoglycosides
Pharmacodynamique
Tétracyclines et aminoglycosides se fixent sur la sous-unité 30S du ribosome, responsable du décodage de l’ARN. Les tétracyclines bloquent la fixation de l’ARNt, empêchant l’élongation de la chaîne protéique : effet bactériostatique. Les aminoglycosides, eux, provoquent une mauvaise lecture de l’ARN, ce qui aboutit à l’incorporation d’acides aminés inadaptés et à la création de protéines défectueuses, parfois intégrées à la paroi cellulaire, causant sa destruction : effet bactéricide.
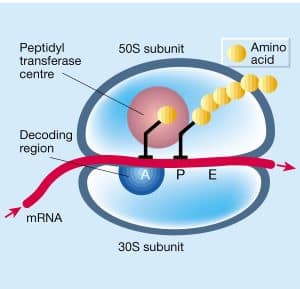
Constante de lipophilité et de dissociation
Le passage des membranes est crucial pour l’efficacité de ces deux familles, car leur cible est intracellulaire. Les tétracyclines, d’une lipophilie modérée et à grand volume de distribution, diffusent bien dans les tissus, notamment les poumons et les mamelles, mais pénètrent peu dans le liquide céphalo-rachidien. Leur absorption digestive est correcte mais diminuée par la présence de nourriture, car ce sont des substances chélatantes.
La doxycycline, plus lipophile, se distingue par une meilleure absorption et des concentrations tissulaires plus élevées. Les aminoglycosides, peu lipophiles et faiblement liés aux protéines plasmatiques, sont mal absorbés par voie orale et agissent surtout dans l’intestin. Après injection, ils atteignent des concentrations efficaces dans différents liquides de l’organisme. Lors d’une inflammation articulaire, la baisse du pH favorise leur diffusion dans les tissus via le piégeage ionique.
Le passage membranaire des tétracyclines dépend de la diffusion, alors que les aminoglycosides nécessitent un transport actif dépendant de l’oxygène, expliquant leur inefficacité contre les bactéries anaérobies. Leur activité diminue également dans les milieux purulents et acides, car les protéines des débris se lient aux aminoglycosides et bloquent leur passage. Dans ce contexte, un drainage des lésions purulentes peut améliorer l’effet de l’antibiotique.
Concentration ou effet dépendant du temps
Les aminoglycosides sont dépendants de la concentration : il faut atteindre des pics élevés sur le site infectieux, tandis que la durée d’exposition importe moins grâce à un effet post-antibiotique prolongé. Un dosage unique quotidien est privilégié pour minimiser le risque de toxicité rénale, car ces molécules s’accumulent dans les cellules du rein. Cette accumulation explique aussi la nécessité d’un délai d’attente prolongé avant l’abattage des animaux traités.
Les tétracyclines n’ont pas de profil strictement dépendant du temps ou de la concentration : l’efficacité repose sur la combinaison des deux paramètres.
Combinaison d’antibiotiques
Combiner des antibiotiques peut parfois élargir le spectre d’activité, mais il faut éviter d’associer inhibiteurs de la synthèse protéique et β-lactamines, car les premiers bloquent la division bactérienne, condition d’efficacité des seconds. Exception faite pour les aminoglycosides, seuls inhibiteurs de synthèse protéique à effet bactéricide, qui peuvent être associés aux β-lactamines pour un effet synergique, notamment contre Streptococcus, Enterococcus, Pseudomonas et d’autres Gram négatif.
Résistance
L’usage massif des tétracyclines a favorisé l’émergence de résistances chez les bovins, porcs et volailles. Plusieurs mécanismes sont impliqués :
- Modification ou protection du site de fixation sur le ribosome
- Diminution de la concentration intracellulaire, par exemple via des pompes à efflux (échange d’un proton contre la tétracycline complexée à un cation) ou une moindre production de porines OPPF, qui servent de passage à travers la membrane externe
- Inactivation enzymatique, surtout chez les entérobactéries
Les pompes à efflux et la modification du ribosome sont les principaux responsables. La résistance croisée est la règle : une souche résistante à une tétracycline l’est généralement aux autres. Pour les aminoglycosides, la résistance découle surtout de l’inactivation enzymatique, mais les autres mécanismes cités pour les tétracyclines existent aussi. La résistance croisée reste toutefois imprévisible et doit être testée au cas par cas.
Macrolides, lincosamides, pleuromutilines et phénicols
Pharmacodynamique
Ces antibiotiques se fixent sur la sous-unité 50S du ribosome, responsable de l’assemblage des acides aminés en chaîne protéique, via la peptidyltransférase. Les lincosamides, pleuromutilines et phénicols bloquent cette enzyme, empêchant la formation de la chaîne. Les macrolides, eux, provoquent la libération prématurée de la protéine en cours d’élongation. Résultat : la bactérie ne produit que des protéines incomplètes ou aucune protéine fonctionnelle.
Ce groupe est classé comme bactériostatique, même si un effet bactéricide apparaît dans certaines conditions (concentration, durée d’exposition, souche, inoculum). Ainsi, le florfénicol a démontré un effet bactéricide sur Actinobacillus pleuropneumoniae et Pasteurella multocida à concentration suffisante pendant 12 heures.
En plus de leur action antibactérienne, certains macrolides et lincosamides exercent des effets immunomodulateurs, notamment ceux à cycle lactonique de 14 ou 15 unités (tels que l’érythromycine). Ces effets nécessitent cependant des concentrations faibles sur une longue durée, ce qui ne s’inscrit pas dans une démarche d’usage raisonné des antibiotiques.
La tilmicosine, par exemple, a fait l’objet d’études montrant une efficacité contre le virus du syndrome reproducteur et respiratoire porcin (SRRP), probablement liée à son accumulation dans les macrophages pulmonaires.
Constante de lipophilité et de dissociation
Le passage membranaire est fondamental pour ce groupe. Leur lipophilie modérée à élevée et leur pKa élevé facilitent la diffusion dans les tissus, y compris les poumons et les mamelles, où le pH est inférieur à celui du plasma (piégeage ionique). Ils atteignent ainsi des concentrations tissulaires supérieures au taux plasmatique, après administration orale ou parentérale. La tulathromycine, par exemple, assure des concentrations élevées et homogènes dans les poumons sur une longue durée, chez plusieurs espèces animales.
Concentration ou effet dépendant du temps
Macrolides, lincosamides, pleuromutilines et phénicols sont typiquement dépendants du temps : il faut maintenir leur concentration au-dessus de la CMI le plus longtemps possible, ce qui justifie une administration continue plutôt que fractionnée.
Combinaison d’antibiotiques
Combiner ces antibiotiques avec des β-lactamines est déconseillé, car leur effet bactériostatique inhibe la division bactérienne, rendant les β-lactamines inefficaces. De plus, ces antibiotiques se fixent sur des sites partiellement similaires sur le ribosome : les associations entre macrolides, lincosamides et phénicols risquent d’être antagonistes. En revanche, une association avec des tétracyclines ou des aminoglycosides reste possible, car ils ciblent une autre sous-unité du ribosome.
Un point de vigilance : ces antibiotiques inhibent les enzymes hépatiques du cytochrome P450, ce qui peut modifier la métabolisation d’autres médicaments et entraîner un surdosage relatif.
Résistance
Chez les bactéries Gram négatif, la résistance aux macrolides, pleuromutilines et lincosamides provient souvent d’une perméabilité membranaire réduite. Chez les Gram positif, elle est due à la modification du site cible, à l’efflux actif ou à l’inactivation enzymatique. Une résistance croisée existe entre macrolides, lincosamides et streptogramines B en raison du chevauchement de leurs sites de fixation (résistance MLSB). Les phénicols sont majoritairement neutralisés par inactivation enzymatique, parfois par efflux actif ou réduction de la perméabilité membranaire. Chez les bactéries multirésistantes, ces mécanismes se combinent.
Références
- Altenburg, J., de Graaf, C.S., van der Werf, T.S., Boersma, W.G. (2011) Effets immunomodulateurs des antibiotiques macrolides, Partie 1 : Mécanismes biologiques. Respiration, 81:67 -74.
- Baert, K., de Baere, S., Croubels, S., Gasthuys, F., de Backer, p. (2001) Pharmacocinétique et biodisponibilité de la sulfadiazine et du triméthoprime (trimazine 30%) après administration orale chez les jeunes porcs non à jeun. J Vet Pharmacol Therap 24:295-298.
- Barberio, A., Badan, M., Bonamico, S., Mancin, M., Simonato, G., Paroline, O. Bazzim, D. (2012) Utilisation du sulfate d’aminosidine pour prévenir la cryptosprodiose chez les veaux. Examen de la médecine vétérinaire 47.
- Benchaoui, H.A., Nowakowski, M., Sherington, J, Rowan, T.G., Sunderland, S.J. (2004) Pharmacocinétique et concentrations de tulathromycine dans les tissus pulmonaires chez les porcs. J.Vet.Pharmacol.Therap 27:203-210.
- Bleyen, N., de Gussem, K., Nguyen, A.D., Ons, E., van Gerven, N. Goddeeris, B. (2009) Effets non curatifs, mais prophylactiques de la paromomycine chez les dindes infectées par Histomonas meleagridis et son effet sur la performance chez les dindes non infectées. Document de recherche, Université de Louvain, Belgique.
- Brouwers, J.R.B.J. (1987) Pharmacocinétique des fluoroquinolones plus récentes. Pharmaceutisch Weekblad Scientifique Edition (Supplément) 9 : S16-S22.
- Brown, M.P., Gronwall, R., Castro, L. (1988) Pharmacocinétique et concentrations de triméthoprim-sulfaméthoxazole dans les juments dans les liquides corporels et endométriaux. Am J Vet Res 49 : 918-922.
- Centre pour la sécurité alimentaire et la santé publique, Université d’État de l’Iowa (2011) Staphylococcus aureus résistant à la méthicilline.
- Fiche d’information du Centre pour la sécurité alimentaire et la santé publique (2011) Staphylococcus aureus résistant à la méthicilline.
- Du, Y., Yoo, D., Paradis, M-A, Scherba, G. (2011) Activité antivirale de la tilmicosine pour le virus du syndrome reproducteur et respiratoire porcin de type 1 et de type 2 dans les macrophages alvéolaires porcins cultivés. Antivir Antirétrovir 3 : 28-33.
- Formuarlium melkvee, werkgroep veterinair antibioticumbeleid KNMVD. Versie 1.1 (2012), aangepast op 02-01-2014.
- Giguere, S., Prescott, J.F., Baggot, J.D., Walker, R.D., Dowling, P.M. (2007) La thérapie antimicrobienne en médecine vétérinaire, quatrième édition. Blackwell édition, 121-137, 179, 188, 191, 205, 207, 229, 231-262.
- Guardabassi, L., Jensen, L.B., en Kruse, H. (2008) Guide de l’utilisation des antimicrobiens chez les animaux. Blackwell Publishing.
- Gustafsson, A., Baverud, V, Franklin, A., Gunnarsson, A., Ögren, G., Ingvast-Larsson, C. (1999) Administration répétée de triméthoprim/sulfadiazine dans la pharmacocinétique du cheval, liaison aux protéines plasmatiques et influence sur la microflore intestinale. J Vet Pharmacol Therap 22:20-26.
- Hamilton-Miller, J.M.T. en Shah, S. (1999) Effet de la concentration d’antibiotiques sur la mort de Staphylococcus aureus et d’Enterococcus faecalis : Comparaison du nouveau penem, Men 10700, avec d’autres antibiotiques β-lactamiques. J Antimicrobes chimiautre 44 : 418-420.
- He, J., Tang, S., Li, L., Zhang, C., Li, X., Xia, Xia, X., Xiao, X. (2010) Pharmacocinétique d’une nouvelle suspension d’amoxicilline/colistine après administration intramusculaire chez des porcs. J vétérinaire Pharmacol Therap 34:42-50.
- Rapport scientifique conjoint de l’ECDC, de l’EFSA et de l’EMEA sur le Staphylococcus aureus résistant à la méthicilline (SARM) chez le bétail, les animaux de compagnie et les aliments.
- Kaartinen, L., Gips, M., Laurila, T., Härtel, H., Soback, S., Pyörälä (2000) Pharmacocinétique de la sulfadoxine et du triméthoprime et irritation des tissus causée par deux produits contenant du sulfate triméthoprime après administration sous-cutanée chez des veaux préruminants. Ré. vétérinaire 31 : 517-526.
- Kaartinen, L., Löhönen, K., Wiese, B., Franklin, A., Pyörälä, S (1999) Pharmacocinétique de sulfadiazine-triméthoprime chez les vaches laitières allaitantes (résumé). Acta Vet Scand 40:271-278.
- Kwiatkowska, B., Maslinska, M., Pryzgodzka, M., Dmowksa-Chalaba, J., Dabrowska, J., Sikorska-Siudek, K. (2013) Système immunitaire comme nouvelle cible thérapeutique pour les antibiotiques. Progrès dans les domaines des biosciences et de la biotechnologie, 4:91-101.
- Leclercq, R., Courvalin, P. (2002) Résistance aux macrolides et aux antibiotiques apparentés chez Streptococcus pneumonie.Agents antimicrobiens et chimiothérapie 46:2727-2734.
- Melchior, M., Van Hout-van Dijk, J. (2011) Antibiotica, van werkingsmechanismen naar antibactériële therapie. Deel III (fourgon IV). Tijdschrift voor Diergeneeskunde 136 (9) : 646-652.
- Mengelers, M.J.B., van Gogh, E.R., Kuiper, H.A. Pijpers, A., Verheijden, J.H.M., van Miert, A.S.J.P.A.M. (1995) Pharmacocinétique de la sulfadiméthoxine et du sulfaméthoxazole en association avec le triméthoprime après administration intraveineuse à des porcs sains et pneumoniques. J Vet Pharmacol Therap 19:243-253.
- Surveillance jaarverslag 2012, Gezondheidsdienst voor dieren te Deventer.
- Nielsen, P., Rasmussen, F. (1975) Triméthoprime et suladoxine chez les porcs ; Demi-vies, volume de distribution et concentration tissulaire. Zbl Vet Med A 22 : 564-571.
- Nielsen, P., Rasmussen, F. (1977) Demi-vie, volume apparent de distribution et liaison aux protéines pour certains sulfamides chez les vaches. Recherche en sciences vétérinaires 22 : 205-208.
- Nielsen, P., Romvary, A., Rasmussen, F. (1978) Sullphadoxine et triméthoprime chez les caprins et les vaches : fraction d’absorption, demi-vie et effet dégradant de la flore ruminale. J Vet Pharmacol Therap 1:37-46.
- Openstax CNX, Diversité probiotique. (bron voor afbeelding).
- Pankey, G.A., Sabath, L.D. (2004) Pertinence clinique du mécanisme d’action bactériostatique par rapport au mécanisme d’action bactéricide dans le traitement des infections bactériennes à Gram positif. Maladies infectieuses cliniques 38 ; pp. 864-870.
- Plumb, D.C. (2011) Manuel des médicaments vétérinaires de Plumb, 7eédition. Wiley-Blackwell.
- Quintiliani, R. Pharmacodynamique des agents antimicrobiens : tuerie dépendante du temps par rapport à la concentration.
- Rasmussen, F., Gelsa, H., Nielsen, P., Pharmacocinétique de la sulfadoxine et du triméthoprime chez les chevaux. Demi-vie et volume de distribution de sulfadoxine et triméthoprime et excrétion cumulative du -triméthoprime. J Vet Pharmacol Therap 2:245-255.
- Rolinski, Z., Duda, M. (1984) Pharmacocinétique analyse du niveau de combinaison sulfamide-triméthoprime chez les veaux (abstrait). Pol J Pharm Pharm 36 : 35-40.
- Romvary, A., Horvay, A. (1976) Données sur la pharmacocinétique des combinaisons sulfonamide-triméthoprime chez les porcs suceurs. Zbl Vet Med 23:781-792.
- Schwarz, S. Chaslus-Dancla, E. (2001) Utilisation d’antimicrobiens en médecine vétérinaire et mécanismes de résistance. Vet Res 32 : 201-225.
- Shoaf, S.E., Schwark, W.S., Guard, C.L. (1989) Pharmacocinétique de la sulfadiazine/triméthoprime chez les veaux mâles néonataux : effet de l’âge et de la pénétration dans le liquide céphalo-rachidien (résumé). Am J Vet Res 50 : 396-403.
- Shoaf, S.E., Schwark, W.S., Guard, C.L., Swartsman, R.V. (1986) Pharmacocinétique de la triméthoprim/sulfadiazine chez les veaux néonatals : influence de la synovite. J Vet Pharmacol Therap 9:446-454.
- van Duijkeren, E., Vulto, A.G., Sloet van Oldruitenborgh, M.M., Kessels, B.G., van Miert, A.S., Breukink, H.J. (1995) Pharmacocinétique de triméthoprime/ sulfachlorpyridazine chez les chevaux après administration orale, nasogastrique et intraveineuse. J Vet Pharmacol Ter 18:47 -53.
- Van Hout-van Dijk, J. et Melchior, M. (2011) Antibiotiques ; des mécanismes d’action à la thérapie antibactérienne. Partie I (de la IV). Journal of Veterinary Medicine 136:494 -499.
- Van Hout-van Dijk, J. et Melchior, M. (2011) Antibiotiques ; des mécanismes d’action à la thérapie antibactérienne. Partie II (de la IV). Journal of Veterinary Medicine 136 (8) : 572-577.
- Van Hout-van Dijk, J. et Melchior, M. (2011) Antibiotiques ; des mécanismes d’action à la thérapie antibactérienne. Partie III (de la IV). Journal de médecine vétérinaire 136:646 -652.
- Van Hout-van Dijk, J. et Melchior, M. (2011) Antibiotiques ; des mécanismes d’action à la thérapie antibactérienne. Partie Lv (de IV). Journal of Veterinary Medicine 136 (10) : 730-733.
- Villarino, N., Brown, S.A., Martin-Jiménez (2013) Comprendre la pharmacocinétique de la tulathromycine : une perspective pulmonaire. Vétérinaire. Pharmacol. Therap. 37:211-221.
- Villarino, N., Lesman, S., Fielder, A., Garciá-Tapia, D., Cox, S., Lucas, M., Robinson, J., Brown, S.A., Martin-Jiménez, T. (2012a) Pharmacocinétique pulmonaire de la tulathromycine chez le porc. Partie I : L’homogénat pulmonaire chez les porcs et les porcs sains a été confronté intratrachéalement au lipopolysaccharide d’Escherichia coli. Vétérinaire. Pharmacol. Therap 36:329-339.
- Villarino, N., Lesman, S., Fielder, A., Garciá-Tapia, D., Cox, S., Lucas, M., Robinson, J., Brown, S.A., Martin-Jiménez, T. (2012b) Pharmacocinétique pulmonaire de la tulathromycine chez le porc. Partie 2 : Compartiments intra-voies respiratoires J. Vet. Pharmacol. Therap 36:340-349.
- Viu, M., Quilez, J., Sanchez-Acedo, C., del Cacho, E., Lopez-Bernad, F. (2000) Essai sur le terrain sur l’efficacité thérapeutique de la paromomycine sur l’infection naturelle à Cryptosporidium parvum dans les labmes. Zoot. Vétérinaire 28 : 13-19.
- Vologodskii, A. (2004) Analyse computationnelle de l’action de l’ADN gyrase. Biophysique journal 87 : 3066-3073.
- Williamson, J.R. (2000) Petite sous-unité, grande science. Biologie moléculaire. Nature 407:306 -307.
Au bout du compte, comprendre les mécanismes d’action, les profils pharmacocinétiques et les stratégies d’association, c’est choisir des antibiotiques avec discernement, limiter la résistance et préserver la santé animale sur le long terme. Le pari de la médecine vétérinaire moderne se joue là, entre vigilance quotidienne et anticipation lucide.



